A hands-on tutorial on simulating tensile fracture in copper using LAMMPS. We compare the simplified Morse potential with the accurate EAM potential and analyze stress-strain behavior during crack propagation.
Understanding how materials fail is fundamental to engineering. Molecular Dynamics (MD) allows us to peek into the atomic scale and observe the precise mechanisms of fracture—something impossible in typical experiments.
In this tutorial, we will verify the behavior of Mode I tensile fracture in a Copper single crystal using LAMMPS. We’ll compare a quick-and-dirty Morse potential against a research-grade EAM potential to see why the choice of physics model matters.
Simulation Preview: Atomic-scale view of Mode I crack propagation in Copper.
In this post, we will:
Derive the Physics: Understand stress, strain, and atomic potentials.
Compare Models: Contrast the simple Morse potential with the accurate Embedded Atom Method (EAM).
Code in LAMMPS: Build complete simulation scripts from scratch.
Analyze Results: Visualize crack propagation and interpret stress-strain curves.
The Physics: Fracture Mechanics at Atomic Scale
Stress and Strain
When a material is pulled, it stretches. At the atomic level, this stretches the bonds between atoms.
$$ \sigma = E \cdot \varepsilon \quad \text{(Elastic Region)} $$
Where:
$\sigma$: Engineering stress (force per unit area)
$E$: Young’s modulus (material stiffness)
$\varepsilon$: Engineering strain (change in length / original length)
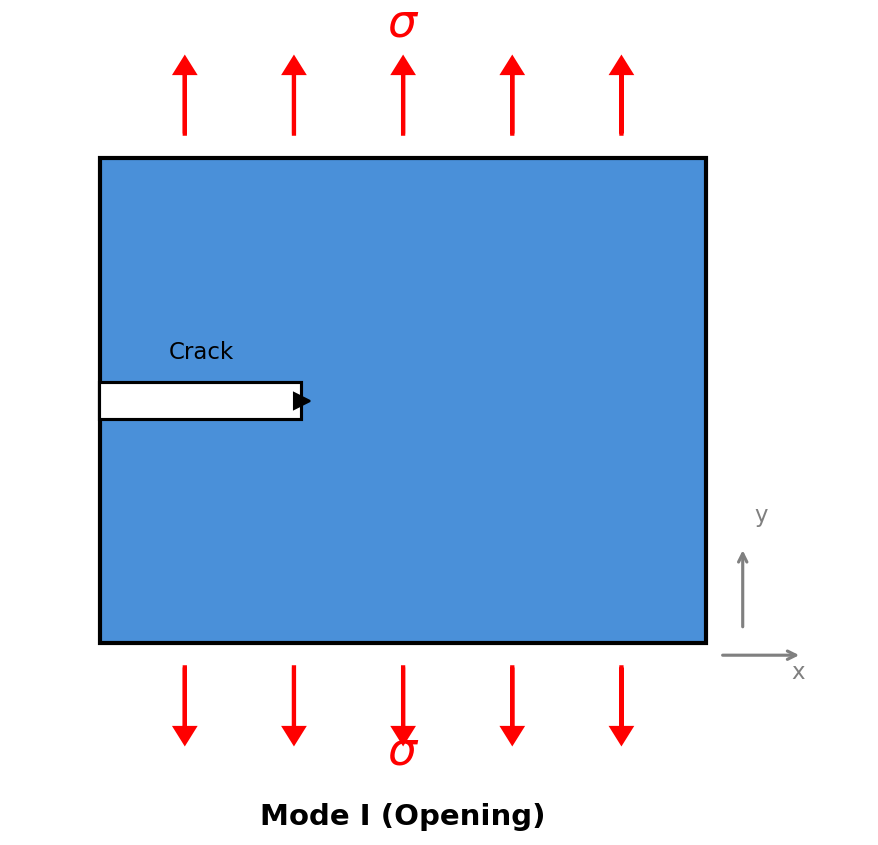
Mode I Fracture
Mode I Fracture
Mode I (Opening Mode) is the most common failure type, where tensile stress pulls the crack faces directly apart. It’s the “tearing” mode of fracture.
Mode I (Opening) Fracture: Tensile stress applied perpendicular to the crack.
At the atomic scale, fracture isn’t just a clean break. It involves:
Bond Stretching: Atoms at the crack tip differ from the bulk.
Bond Breaking: When local stress exceeds bond strength.
Dissipation: Energy loss through heat or plastic deformation (dislocation movement).
Interatomic Potentials: Morse vs EAM
The “brain” of any MD simulation is the interatomic potential—the mathematical rule describing how atoms interact. We’ll compare two vastly different approaches:
Why the Choice Matters
Pair Potentials (e.g., Morse): Assume interaction depends only on the distance between two atoms. Simple, but ignores the “electron glue” effect in metals.
Many-Body Potentials (e.g., EAM): Account for the local electron density. An atom “feels” difference based on how many neighbors it has. This is critical for surfaces and cracks.
Morse Potential (Simplified)
The Morse potential is a computationally cheap pair potential:
# ==============================================================================# Metal Fracture Simulation - Full Version (EAM Potential)# ==============================================================================# Uses EAM potential for accurate copper properties.# Requires: Cu_mishin1.eam.alloy (download from NIST potentials repository)## Author: MD Tutorial# Units: Metal units (Angstroms, eV, ps, K)# ==============================================================================# Create output directoryshellmkdir-pfracture_results_eam# =====================# 1. Initialization# =====================unitsmetaldimension3boundarypsp# Periodic in x,z; free surface in yatom_styleatomic# =====================# 2. Create Copper Crystal (FCC)# =====================latticefcc3.615orientx100orienty010orientz001# Larger box: ~100 x 50 x 14 Angstroms (~8000 atoms)regionboxblock02801404unitslatticecreate_box1boxcreate_atoms1box# =====================# 3. Create Pre-existing Crack# =====================regioncrackblock096.57.5INFINFunitslatticedelete_atomsregioncrackvariablenatomsequalcount(all)print"Atoms after crack: ${natoms}"# =====================# 4. EAM Potential for Copper# =====================# Mishin EAM potential - accurate for Cu mechanical properties# Download from: https://www.ctcms.nist.gov/potentials/pair_styleeam/alloypair_coeff**Cu_mishin1.eam.alloyCu# =====================# 5. Define Regions and Groups# =====================regiontop_gripblockINFINF13INFINFINFunitslatticeregionbot_gripblockINFINFINF1INFINFunitslatticegrouptop_atomsregiontop_gripgroupbot_atomsregionbot_gripgroupboundaryuniontop_atomsbot_atomsgroupmobilesubtractallboundary# =====================# 6-12. Same as Morse version...# =====================# (See full script in repository)run50000
# Run simplified version (no dependencies)lmp-inmetal_fracture_simple.in# Run full version (requires EAM file)lmp-inmetal_fracture.in
Results and Analysis
Stress-Strain Curves
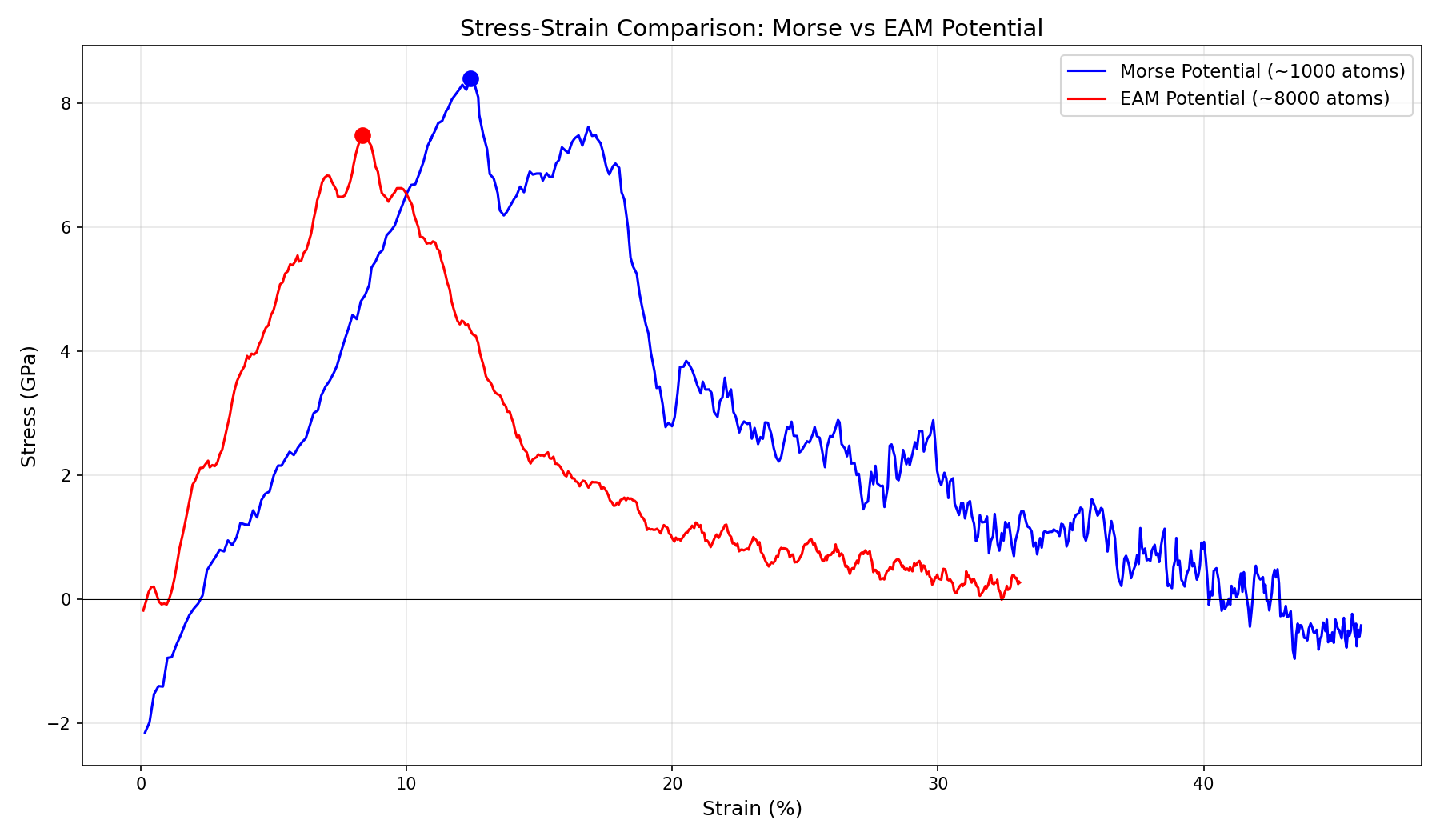
Stress-Strain Comparison: Morse vs EAM Potential
The stress-strain curves reveal distinct mechanical behaviors:
Property
Morse Potential
EAM Potential
Peak Stress
~8.4 GPa
~7.5 GPa
Strain at Peak
~12%
~8%
Post-Peak Behavior
Gradual decline
Sharp drop
Failure Mode
Ductile-like
Brittle-like
Visualizing Crack Propagation
Since the stress-strain curves suggest different failure modes, let’s look at the atomic trajectories.
Morse Potential: Ductile hole growth
EAM Potential: Sharp brittle crack
Key Observations
Peak Strength: Both predict ~7-8 GPa. This is theoretically reasonable for a flawed crystal at these high strain rates, but much higher than bulk experimental copper.
Failure Strain: Morse stretches to ~12% strain, behaving like “gum”. EAM snaps at ~8%, capturing the brittle nature of high-speed fracture.
Mechanism: EAM accurately captures the surface energy cost of creating a new crack surface. Morse underestimates this, allowing the material to resist fracture longer.
Snap-back: The sharp drop in the EAM curve (Brittle) vs the gradual decline in Morse (Ductile) is the signature difference.
Summary and Recommendations
When to Use Each Potential
Use Case
Recommended Potential
Learning/Testing
Morse
Quick prototyping
Morse
Qualitative trends
Morse
Publication-quality
EAM
Quantitative predictions
EAM
Multi-element systems
EAM/MEAM
Key Takeaways
Potentials are Physics: They aren’t just parameters. Morse gives you “balls on springs”. EAM gives you “atoms in an electron sea”.
Surface Energy Drives Fracture: Creating a crack means creating surface. If your potential gets surface energy wrong (like Morse), your fracture mechanics will be wrong.
Timescales Matter: MD happens in picoseconds. The strain rates ($10^9 s^{-1}$) are explosive. Don’t expect to match macroscopic tensile test numbers directly.
References
Mishin, Y., et al. “Structural stability and lattice defects in copper: Ab initio, tight-binding, and embedded-atom calculations.” Physical Review B 63.22 (2001): 224106.
Farkas, D. “Atomistic simulations of metallic microstructures.” Current Opinion in Solid State and Materials Science 17.6 (2013): 284-297.