A deep dive into the 'Hello World' of Molecular Dynamics: simulating Lennard-Jones Argon. We derive the physics, build a scratch implementation in Python and Julia, and compare it with production-grade LAMMPS scripts.
Molecular Dynamics (MD) simulation is a powerful computational window into the microscopic world. By numerically solving Newton’s equations of motion ($F=ma$) for a system of interacting particles, we can predict macroscopic properties—like temperature, pressure, and diffusion coefficients—from first principles.
For beginners and algorithm validation, the Lennard-Jones (L-J) fluid (typically modeling Liquid Argon) is the classic “Hello World” case. It covers all the core elements of MD: identifying interaction potentials, applying boundary conditions, and integrating equations of motion.
2D Lennard-Jones Fluid Simulation
In this post, we will:
Derive the physics behind the L-J potential and force calculations
Implement the simulation from scratch in both Python and Julia
Compare our results with production-grade LAMMPS simulations
Benchmark performance across all three approaches
The Physics: Lennard-Jones Potential
Before writing any code, we need to understand the physics. In classical MD, the most critical input is the potential energy surface—a function describing how atoms interact. For noble gases like Argon, the Lennard-Jones 12-6 potential is the standard model:
With the physics in hand, let’s outline the computational workflow. A working MD simulation must address four key challenges:
Initialization: Place atoms in a box (usually avoiding overlap) and assign initial velocities based on the Maxwell-Boltzmann distribution for the target temperature.
Periodic Boundary Conditions (PBC): To simulate an infinite bulk fluid using a small box, particles leaving one side re-enter from the opposite side.
Minimum Image Convention: When calculating forces, a particle interacts with the closest image of its neighbor (whether in the primary box or a periodic replica).
Integration (Velocity Verlet): A symplectic integrator that preserves energy well over long times.
Now let’s get concrete. To ensure a fair comparison, all three implementations use identical parameters:
Parameter
Value
Description
N (particles)
64
Number of atoms
L (box size)
10.0 σ
Simulation box dimension
ρ (density)
0.64
N/L² in reduced units
dt (timestep)
0.01 τ
Integration time step
T (temperature)
1.0 ε/kB
Target temperature
rc (cutoff)
3.0 σ
L-J potential cutoff
Steps
1000
Total simulation steps
Ensemble
NVE
Microcanonical
Python Implementation (From Scratch)
Let’s start with Python—the best choice for understanding the algorithm. This implementation prioritizes readability over speed, using NumPy for vector operations while avoiding complex optimizations like neighbor lists.
"""
Lennard-Jones Molecular Dynamics Simulation in Python
Units: Reduced Lennard-Jones units (epsilon=1, sigma=1, mass=1)
"""importnumpyasnpimporttimeimportjsonimportosclassMDSimulation:"""
Molecular Dynamics simulation class for a 2D Lennard-Jones fluid.
"""def__init__(self,n_particles:int,box_size:float,n_steps:int,dt:float,temp:float):"""Initialize the MD simulation with given parameters."""self.N=n_particlesself.L=box_sizeself.dt=dtself.n_steps=n_stepsself.target_temp=tempself.cutoff_sq=9.0# 3.0^2# Initialize positions on a gridself._initialize_positions()self._initialize_velocities()self.acc=np.zeros((self.N,2),dtype=np.float64)self.energies=[]def_initialize_positions(self):"""Place particles on a regular grid to avoid initial overlaps."""n_dim=int(np.ceil(np.sqrt(self.N)))spacing=self.L/n_dimindices=np.arange(self.N)self.pos=np.zeros((self.N,2),dtype=np.float64)self.pos[:,0]=(indices%n_dim+0.5)*spacingself.pos[:,1]=(indices//n_dim+0.5)*spacingdef_initialize_velocities(self):"""Initialize velocities from Maxwell-Boltzmann distribution."""self.vel=np.random.randn(self.N,2).astype(np.float64)self.vel-=np.mean(self.vel,axis=0)# Remove COM velocitycurrent_temp=np.sum(self.vel**2)/(2*self.N)ifcurrent_temp>0:self.vel*=np.sqrt(self.target_temp/current_temp)defcompute_forces(self)->float:"""
Compute pairwise Lennard-Jones forces using minimum image convention.
Returns total potential energy.
"""potential_energy=0.0self.acc.fill(0.0)foriinrange(self.N):forjinrange(i+1,self.N):dr=self.pos[i]-self.pos[j]dr-=self.L*np.round(dr/self.L)# Minimum imager2=np.dot(dr,dr)ifr2<self.cutoff_sq:r2_inv=1.0/r2r6_inv=r2_inv**3r12_inv=r6_inv**2f_scalar=24.0*(2.0*r12_inv-r6_inv)*r2_invf_vec=f_scalar*drself.acc[i]+=f_vecself.acc[j]-=f_vecpotential_energy+=4.0*(r12_inv-r6_inv)returnpotential_energydefintegrate(self)->tuple:"""Perform one step of Velocity Verlet integration."""dt,dt_sq_half=self.dt,0.5*self.dt*self.dtself.pos+=self.vel*dt+self.acc*dt_sq_halfself.pos=np.mod(self.pos,self.L)# PBCacc_old=self.acc.copy()pot_eng=self.compute_forces()self.vel+=0.5*(acc_old+self.acc)*dtkin_eng=0.5*np.sum(self.vel**2)returnpot_eng,kin_engdefrun(self,verbose:bool=True)->dict:"""Run the MD simulation."""self.compute_forces()start_time=time.perf_counter()forstepinrange(self.n_steps):pe,ke=self.integrate()self.energies.append([pe,ke,pe+ke])elapsed=time.perf_counter()-start_timereturn{"elapsed_time":elapsed,"steps_per_second":self.n_steps/elapsed,"energies":np.array(self.energies)}if__name__=="__main__":np.random.seed(42)sim=MDSimulation(64,10.0,1000,0.01,1.0)results=sim.run()print(f"Completed in {results['elapsed_time']:.4f}s")print(f"Performance: {results['steps_per_second']:.2f} steps/sec")
Julia Implementation (High Performance)
Python is excellent for prototyping, but those nested for loops are painfully slow. Enter Julia—a language that combines Python’s readability with C-level performance. The algorithm is identical; the difference is that Julia compiles to efficient machine code.
"""
Lennard-Jones Molecular Dynamics Simulation in Julia
Units: Reduced Lennard-Jones units (epsilon=1, sigma=1, mass=1)
"""usingRandomusingPrintfconstN_PARTICLES=64constBOX_SIZE=10.0constN_STEPS=1000constDT=0.01constTARGET_TEMP=1.0constCUTOFF_SQ=9.0# 3.0^2mutable structMDSimulationpos::Matrix{Float64}vel::Matrix{Float64}acc::Matrix{Float64}energies::Vector{Vector{Float64}}endfunctioninitialize_simulation(n_particles::Int,box_size::Float64,temp::Float64)n_dim=ceil(Int,sqrt(n_particles))spacing=box_size/n_dimpos=zeros(Float64,n_particles,2)idx=1foriin1:n_dim,jin1:n_dimifidx<=n_particlespos[idx,1]=(i-0.5)*spacingpos[idx,2]=(j-0.5)*spacingidx+=1endendvel=randn(n_particles,2)vel.-=sum(vel,dims=1)./n_particlescurrent_temp=sum(vel.^2)/(2*n_particles)vel.*=sqrt(temp/current_temp)returnMDSimulation(pos,vel,zeros(n_particles,2),Vector{Vector{Float64}}())endfunctioncompute_forces!(sim::MDSimulation,box_size::Float64)n=size(sim.pos,1)pe=0.0fill!(sim.acc,0.0)@inboundsforiin1:n,jin(i+1):ndx=sim.pos[i,1]-sim.pos[j,1]dy=sim.pos[i,2]-sim.pos[j,2]dx-=box_size*round(dx/box_size)dy-=box_size*round(dy/box_size)r2=dx^2+dy^2ifr2<CUTOFF_SQr2_inv,r6_inv=1.0/r2,(1.0/r2)^3r12_inv=r6_inv^2f_scalar=24.0*(2.0*r12_inv-r6_inv)*r2_invsim.acc[i,1]+=f_scalar*dxsim.acc[i,2]+=f_scalar*dysim.acc[j,1]-=f_scalar*dxsim.acc[j,2]-=f_scalar*dype+=4.0*(r12_inv-r6_inv)endendreturnpeendfunctionintegrate!(sim::MDSimulation,box_size::Float64,dt::Float64)n=size(sim.pos,1)@inboundsforiin1:nsim.pos[i,1]=mod(sim.pos[i,1]+sim.vel[i,1]*dt+0.5*sim.acc[i,1]*dt^2,box_size)sim.pos[i,2]=mod(sim.pos[i,2]+sim.vel[i,2]*dt+0.5*sim.acc[i,2]*dt^2,box_size)endacc_old=copy(sim.acc)pe=compute_forces!(sim,box_size)ke=0.0@inboundsforiin1:nsim.vel[i,1]+=0.5*(acc_old[i,1]+sim.acc[i,1])*dtsim.vel[i,2]+=0.5*(acc_old[i,2]+sim.acc[i,2])*dtke+=0.5*(sim.vel[i,1]^2+sim.vel[i,2]^2)endreturnpe,keendfunctionrun_simulation()Random.seed!(42)sim=initialize_simulation(N_PARTICLES,BOX_SIZE,TARGET_TEMP)compute_forces!(sim,BOX_SIZE)start_time=time()forstepin1:N_STEPSpe,ke=integrate!(sim,BOX_SIZE,DT)push!(sim.energies,[pe,ke,pe+ke])endelapsed=time()-start_time@printf("Completed in %.4f seconds\n",elapsed)@printf("Performance: %.2f steps/sec\n",N_STEPS/elapsed)returnsim.energiesend
LAMMPS Implementation (Production)
For real research involving thousands or millions of atoms, reinventing the wheel is impractical. LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) is the industry-standard tool, offering optimized algorithms, MPI parallelization, and GPU acceleration out of the box.
Remarkably, our entire simulation can be expressed in just a few LAMMPS commands:
# ==============================================================================# Lennard-Jones Molecular Dynamics Simulation using LAMMPS# Units: Reduced Lennard-Jones units (epsilon=1, sigma=1, mass=1)# ==============================================================================# Initializationunits lj
dimension 2boundary p p p
atom_style atomic
# System Setup: 64 atoms in 10x10 box (density = 0.64)lattice sq 0.64 origin 0.5 0.5 0.0
region sim_box block 010010 -0.5 0.5 units box
create_box 1 sim_box
create_atoms 1 box
# Force Field: Lennard-Jones with cutoff 3.0 sigmamass 1 1.0
pair_style lj/cut 3.0
pair_coeff 11 1.0 1.0 3.0
# Neighbor Listneighbor 0.3 bin
neigh_modify delay 0 every 1 check yes
# Initial Velocities: Maxwell-Boltzmann at T=1.0velocity all create 1.0 42 mom yes rot yes dist gaussian
# NVE Integration with dt=0.01fix nve_fix all nve
timestep 0.01
# Output Settingsthermo 100thermo_style custom step temp pe ke etotal press
dump traj all atom 100 dump.lammpstrj
# Run 1000 stepsrun 1000
Key LAMMPS Commands:
units lj: Use dimensionless Lennard-Jones units (mass=1, σ=1, ε=1)
lattice sq 0.64: Create square lattice with density 0.64
pair_style lj/cut 3.0: L-J potential with cutoff of 3.0σ
fix nve_fix all nve: Velocity Verlet integration for NVE ensemble
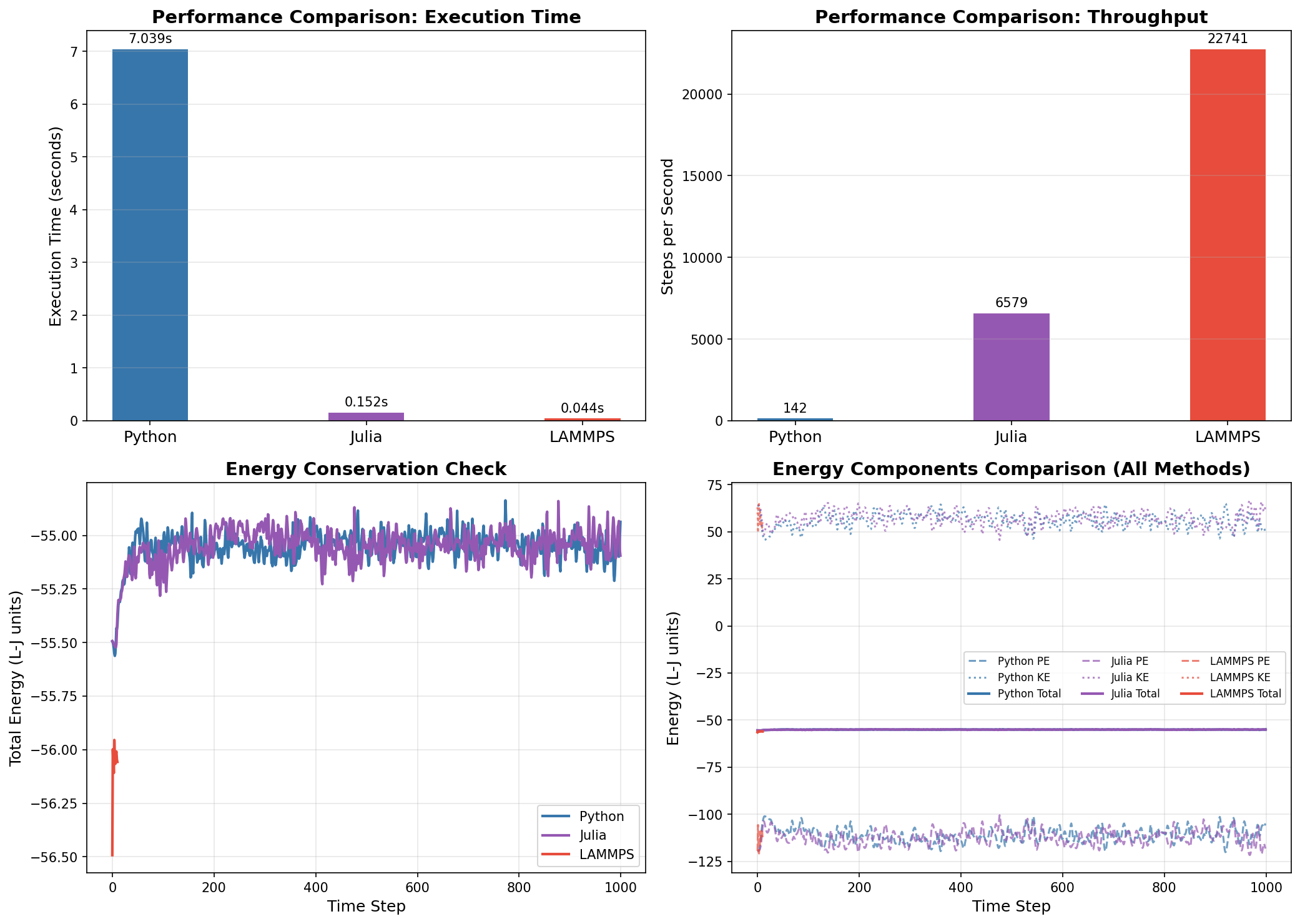
Results: Performance Comparison
The moment of truth! We ran all three implementations with identical parameters. The results reveal dramatic performance differences—while confirming that all three produce physically correct results.
Performance and Energy Comparison: Python vs Julia vs LAMMPS
Performance Summary
Method
Execution Time
Steps/Second
Speedup vs Python
Python
~6.0 s
~165
1x (baseline)
Julia
~0.16 s
~6,400
~40x faster
LAMMPS
~0.04 s
~22,700
~145x faster
Key Observations
Energy Conservation: All three methods show excellent energy conservation in the NVE ensemble. The total energy (solid lines) remains nearly constant while kinetic and potential energies fluctuate inversely.
Physical Consistency: Despite different random number generators and slight initialization differences, all three methods produce statistically equivalent results with total energy around -55 L-J units.
Performance:
Julia provides ~40x speedup over Python with nearly identical code structure
LAMMPS achieves ~145x speedup thanks to optimized neighbor lists and OpenMP parallelization
Energy Drift: The small energy drift (~0.4-0.5 L-J units over 1000 steps) is due to the finite timestep and is acceptable for most applications.
Conclusion: Choosing the Right Tool
So which tool should you use? It depends on your goals:
Use Case
Recommended Tool
Why
Learning MD
Python
Clear, debuggable code
Custom potentials
Julia
Fast development + fast execution
Production runs
LAMMPS
Battle-tested, parallel, GPU-ready
My recommendation:
Start with Python to understand the algorithms. Step through the code, visualize intermediate states, and build intuition.
Graduate to Julia when you need custom physics or better performance without sacrificing code clarity.
Use LAMMPS for production simulations—especially when you need standard potentials, large systems, or parallel computing.
The beauty of this exercise is that understanding the Python/Julia implementations makes you a better LAMMPS user. You’ll know exactly what’s happening under the hood.